
Systemic inflammation drives brain neurodegeneration
 In a richly valuable paper published recently in Frontiers in Cellular Neuroscience the authors describe the ways in which systemic inflammation causes neurodegeneration in the brain associated with cognitive decline and a host of neuropsychiatric disorders. In the short term this manifests the anorexia, malaise, depression, and decreased physical activity known as sickness behavior (SB) that occurs with inflammation due to infection. Permanent cognitive and behavioral changes due to neurodegeneration occur when inflammation is chronic. Discerning and targeting the causes of inflammation offers opportunities for treatment.
In a richly valuable paper published recently in Frontiers in Cellular Neuroscience the authors describe the ways in which systemic inflammation causes neurodegeneration in the brain associated with cognitive decline and a host of neuropsychiatric disorders. In the short term this manifests the anorexia, malaise, depression, and decreased physical activity known as sickness behavior (SB) that occurs with inflammation due to infection. Permanent cognitive and behavioral changes due to neurodegeneration occur when inflammation is chronic. Discerning and targeting the causes of inflammation offers opportunities for treatment.
Neuroimmune modulation
The nervous system senses inflammation directly and can exert control through the vagus nerve:
"The efferent axis of neuroimmune control is better understood after the cholinergic anti-inflammatory pathway (CAP), a cholinergic reflex system that regulates inflammation via the vagus nerve that stimulates the splenic nerve to release noradrenaline. Noradrenaline in turn stimulates a subset of acetylcholine (ACh)-producing splenic T-cells (CD4+CD44hiCD62Llo) to release ACh, which binds to α7 nicotinic receptors on the surface of macrophages, resulting in down-regulation of TNF by blocking the nuclear translocation of nuclear factor kappa B (NF-κB). Thus far, this is a unique scenario in which an immune cell acts as interneuron in a reflex system. Electrical as well as chemical stimulation of the CAP have been shown to decrease the inflammatory burden and increase survival of experimental sepsis."
The a cholinergic response expressed through the vagus nerve can wind down inflammation and protect against neurodegeneration.
Sickness behavior
Transient inflammation, such as associated with a cold or flu, produces behavioral symptoms of the same character as those which persist with the chronic systemic inflammation that can drive neurodegeneration.
"The acute effects of systemic inflammation upon cognition and behavior are not limited to the elderly or the critically ill. As we have witnessed in ourselves and those near us, even a minor and self-limited common cold induces a transient syndrome known as sickness behavior (SB) marked by fatigue, depression, lack of drive, malaise, sleep disturbances, decreased physical activity, and social interactions, as well as cognitive impairment. Healthy volunteers develop anxiety, depression, and memory impairment in response to a low dose of lipopolysaccharide (LPS), and the development of such clinical scenario correlates with TNF secretion."
And patients with chronic infections such as tuberculosis, human immunodeficiency virus (HIV), hepatitis B virus (HVB), and hepatitis C virus (HCV) can have cognitive and behavioral problems due to the persistent inflammatory response.
"This supports the role of large loads of inflammatory cytokines in inducing and sustaining brain dysfunction. Experimentally, NADPH oxidative activity and nitric oxide synthase (iNOS) are induced in the brain shortly after systemic inflammation, potentially leading to NMDA-dependent neurotoxicity"
Sepsis and severe trauma
An overwhelming load of pathogens or severe trauma can unleash an immune inflammatory response that results in neurodegeneration.
"Under normal conditions, inflammation is a well-orchestrated response with constant fine-tuning. Once microorganisms have breached the skin and mucosal barriers, innate immunity is critical in preventing further invasion by launching inflammation. After the infection source has been cleared, the inflammatory response also plays an important role in tissue repair and functional healing. When the source of damage has been controlled, the same mechanisms that initiated and regulated inflammation will dampen the response. Large loads of pathogens, or infection by highly virulent pathogens, can trigger an en-masse systemic response that leads to sepsis and multiple organ failure...The nervous system is particularly vulnerable to damage in response to systemic inflammation."
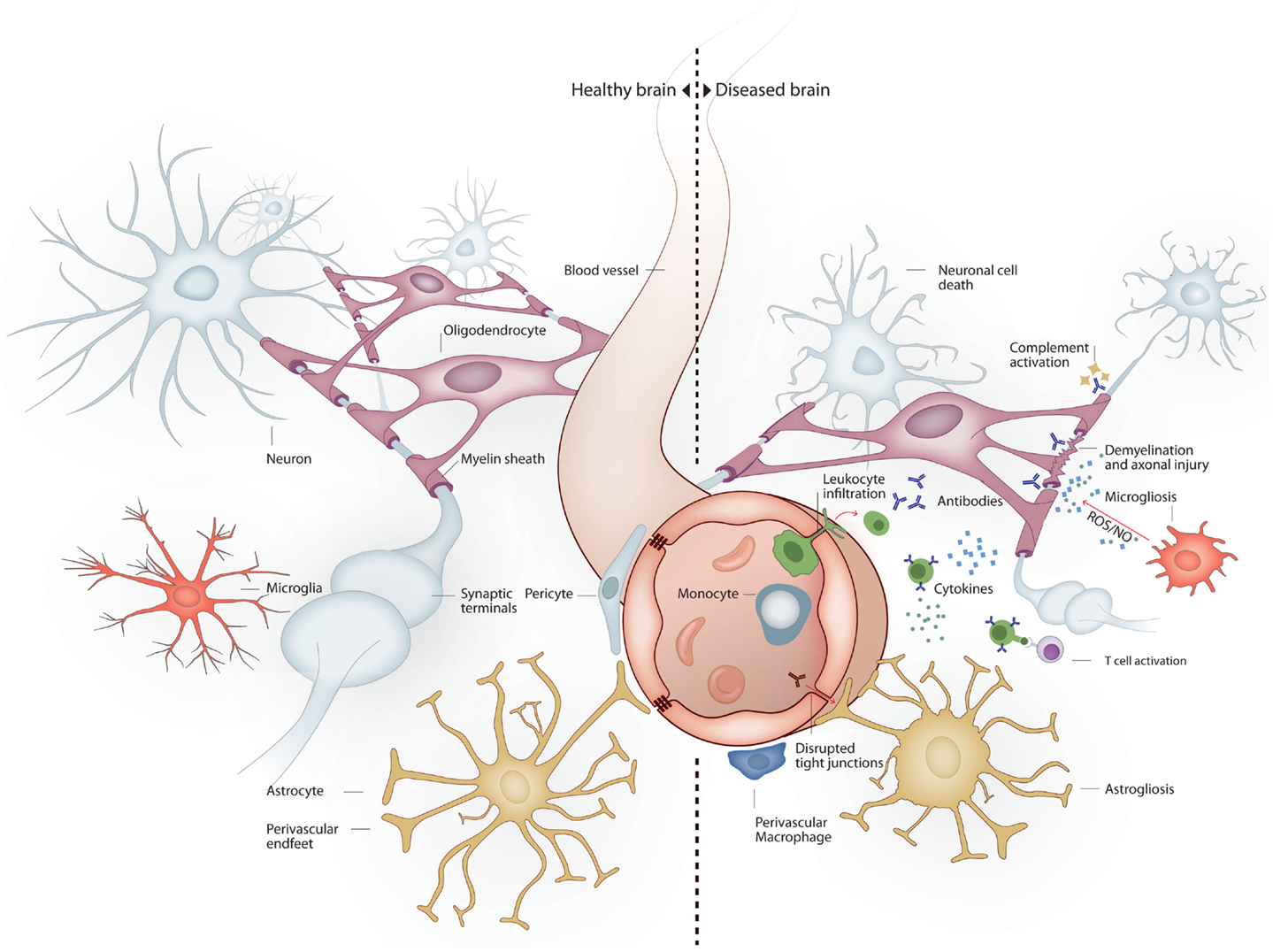
"Inflammation-induced infiltration of immune cells and mediators into the brain leads to profound structural and functional changes. As a consequence, up to 81% of septic patients develop sepsis-associated delirium (SAD), with elderly patients being at particularly high risk. In the elderly, severe sepsis is sufficient to trigger new cognitive decline of sufficient importance as to profoundly interfere with quality of life...Neonatal sepsis is also marked by abnormalities of the white matter (66% of infants in one cohort), and white matter lesions correlate to poorer mental and psychomotor development at 2 years.
Moreover...
"...clearing the trigger of sepsis does not prevent the appearance of persistent brain damage...in a model of endotoxemia in aged rats, a single systemic injection of LPS induced brain inflammation that lasted for at least 30 days...This suggests that even transient bouts of systemic inflammation of only limited significance can cause sustained brain damage."
Traumatic inflammation also promotes neurodegeneration:
"Severe trauma, as well as surgery can lead to large loads of endogenous pro-inflammatory molecules (damage-associated molecular patterns (DAMPs) being released. A few DAMPs have been shown to induce brain dysfunction in vivo. Of those, TNF and IL-1 can mediate long-standing cognitive and behavioral changes and, in experimental settings, interfering with the effect of TNF reduces the effect of trauma in the formation of contextual memory."
In this context antioxidants can have neuroprotective effects.
"Experimentally, preemptive administration of the free radical scavenger endarvone before sepsis induction resulted in reduced neuronal damage and blood–brain barrier (BBB) permeability. Administration of the antioxidants N-acetylcysteine and deferoxamine shortly after murine sepsis induction has shown long-term neuroprotective effects."
Systemic inflammation disrupts brain networks
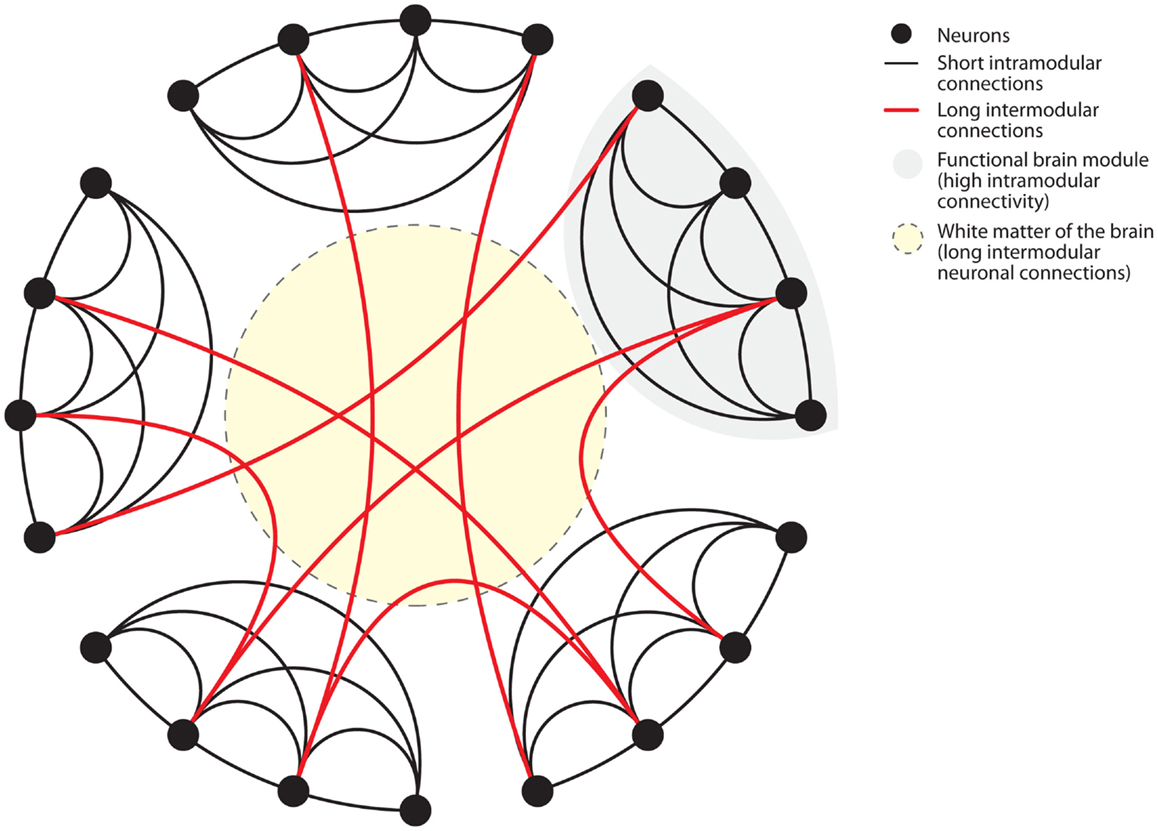 The brain is characterized as a 'small-world' network with two levels of connection that are susceptible to disruption by inflammation.
The brain is characterized as a 'small-world' network with two levels of connection that are susceptible to disruption by inflammation.
"Biological systems, such as the neuronal network of the human brain have “small-world” properties. Small-world networks have two levels of organization. On the local level, groups of neurons specialized in a specific task form functional modules with high short intramodular connectivity. On the global level, different modules are connected through long intermodular connections. The advantage of the latter type of connections is enhanced computational efficiency through parallel processing of information. Anatomically, long intermodular connections are formed by axonal fiber tracts in the white matter. Long fibers are characterized by high energetic “wiring costs”. To provide the energy for the maintenance of these long fibers the brain is relying on a constant energy supply. Recent findings have elegantly identified oligodendrocyte-derived lactate as the main energetic substrates for axonal maintenance. Consistently, disruption of this oligodendrocyte-neuronal metabolic coupling triggered neurodegeneration. Systemic inflammation poses dramatic challenges to the energetic supply of the brain."
The brain requires a constant stream of nutrients to maintain its 'wiring'
Autoimmune driven neuroinflammation, among other insults, can disrupt the delivery of nutrients to neurons and contribute to mitochondrial dysfunction.
"To cover its wiring costs the brain is highly reliant on a constant nutrient supply. Nutrient supply through blood vessels can be compromised through vascular pathologies associated with systemic inflammation...Autoimmune disorders have a chronic course of vascular pathology with acute flares. The most common vascular pathology is the autoantibody-associated antiphospholipid syndrome. Patients with antiphospholipid syndrome display cognitive deficits. MRI studies found diffuse infarctions and white matter lesions in these patients...In line with the concept of high “wiring costs” imposed on the brain by long intermodular connections,Hans Lassmann argues that inflammation in MS causes mitochondrial damage and inability of the brain to maintain neuronal processes. The source of mitochondrial damage is radicals formed as a consequence of inflammation in MS."
Energy crisis for the brain
Systemic inflammation damages connectivity and fuels mitochondrial dysfunction...
"Taken together these findings indicate that systemic inflammation leads to an energy crisis of the brain that reduces its connectivity. Oxidative stress might be the main mediator of this pathology. Thus, inflammation-induced changes in the brain resemble hallmarks of the aged brain where oxidative damage leads to decreased expression of genes associated with synaptic plasticity and increased expression of stress-response genes. Likewise, the brain during systemic inflammation shows hallmarks of neurodegenerative diseases where oxidative stress and mitochondrial damage have consistently been found."
Balancing act
Normally astrocytes and neurons talk to each other to keep activation of brain immune cells in check. But this can get out of hand in response to a pathogen resulting in serious damage. Balance is maintained, partly by accepting a certain degree of tolerance for pathogens:
"Brain-resident microglia and peripheral immune cells maintain immune surveillance of brain parenchyma, CSF, and perivascular space for infectious agents or damage-associated milieu changes. In the case of brain infection, complete eradication of some invading pathogens can only be achieved at the cost of irreparable damage to brain tissue. To prevent such damage, the immune system has established active mechanisms of pathogen tolerance. Examples for coexistence-prone pathogens are herpes simplex virus type I or Cryptococcus gattii. A growing body of evidence indicates that not only immune tolerance but also resolution of neuroinflammation is a tightly regulated active immunological process. Taken together, anti-inflammatory brain milieu, pathogen tolerance, and resolution of neuroinflammation require a balanced action between different branches of the immune system."
Dysregulation causing systemic inflammation drives neurodegeneration
There are a number of mechanisms by which dysregulated systemic inflammation promotes neuroinflammation and neurodegeneration. These in include activation of apoptosis through the inflammasome (inflammation signalling chains):
"Apoptosis is one of the main drivers of neurodegeneration. Apoptosis and cell death constantly occur under physiological conditions throughout the human body and cell debris is cleared by immune cells mostly without induction of chronic inflammation. However, during systemic inflammation, apoptosis of stressed cells might further exacerbate the underlying pathology. Activators of apoptosis lead to direct or indirect activation of caspases...inflammatory caspases are crucial for the activation of the innate immune system through the inflammasome...Activation of the innate immune system through the inflammasome is a driver of pathology in age-associated and autoimmune neurodegenerative disorders...these finding show an intricate relationship between inflammation and activation of apoptosis"
Microvesicles (MVs) packed with inflammatory messengers are secreted by peripheral and brain immune cells contribute to neurodegeneration:
"Cellular components of innate immunity can pack and secrete inflammatory messengers in microvesicles (MVs). Peripheral macrophages, as well as brain microglia can secrete inflammasome components (caspase-1, IL-1β, and IL-18) in MVs, and the presence of extravesicular inflammatory inducers (e.g., astrocitic ATP) is sufficient to induce the neurotoxicity by the inflammatory load of MVs."
This correlates with disease activity in multiple sclerosis and also a significant role in Alzheimer's disease (AD):
"Recent evidence suggest that MVs play a critical role in the spectrum of AD as well. MVs released by activated microglia participate in the neurodegenerative process of AD by promoting the generation of highly neurotoxic soluble forms of β-amyloid. Based on this collective evidence, it is now clear that EVs produced by peripheral myeloid cells, as well as immune brain cells, are novel and potentially critical biomarkers for neuroinflammatory conditions by providing a link between inflammation and neurodegeneration."

Immune cells and mediators drive neurodegeneration
Immune cells in both the periphery and the brain can cause neuronal apoptosis through multiple pathways that can be targeted for therapy:
"Various triggers of apoptosis have been described with respect to the brain. Neuronal apoptosis can be directly induced by ROS, pro-inflammatory cytokines or activated immune cells...Additionally, damaged mitochondria are a major source of ROS and mediators of apoptosis. Conversely, inactivation of ROS has anti-apoptotic effects. The inflammatory cytokine TNFα and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) directly induce neuronal apoptosis. Additionally, intracerebroventricularly injected TNFα was shown to induce depression-like symptoms. Cytokine mediated induction of apoptosis was also observed by IL-1β."
Immune cells in the brain and the periphery cause neurodegeneration, with evidence that antiinflammatory interventions can oppose neuronal death.
"Sources of cytokines under systemic inflammation are brain resident, paravascular or peripheral immune cells. Furthermore, activated immune cells can directly induce neuronal cell death. Brain-resident microglia convey neuronal toxicity through various mechanisms including secretion of neurotoxic factors, as well as through activation of cyclooxygenase/prostaglandin E2 (COX/PGE2) pathways. In fact, blocking the COX/PGE2 pathway by experimentally deleting the prostaglandine receptor EP2 increases mitochondrial degradation of β-amyloid, potentially opening a new therapeutic avenue for AD."
When there is systemic inflammation immune cells in the periphery in the body can gain access to the brain through the blood-brain barrier:
"Peripheral immune cells can penetrate the BBB under conditions of systemic inflammation and contribute to brain pathology. Cytotoxic T-cells were shown to be directly neurotoxic in autoimmune and aging-associated neurodegenerative disorders of the CNS. Co-localization of T-cells with neurons and neuron-specific cytotoxicity of T-cells was shown in vivo and in vitro."
Anti-brain antibodies*
Identification of anti-brain antibodies is a key clinical finding that practitioners in a wide range of disciplines should be alert for.
"B-cell-derived anti-brain antibodies have been identified as drivers of brain pathology in various diseases. In the last decade, an increasing number of anti-brain antibodies has been detected that can affect cognition and behavior...Under pathological conditions, antibodies may penetrate the BBB through different mechanisms including local and systemic inflammation, or antigen mediated endocytosis."
Anti-NMDA antibodies have been receiving much scrutiny for neuropsychiatric and neurodegenerative disorders.
"Furthermore, NMDA-receptor-specific antibodies to the subunit 2 (GluN2) have been found in a subset of SLE patients with neuropsychiatric symptoms. These antibodies are cross-reactive to DNA...DNA–NMDA receptor antibodies preferentially bind the open configuration of the NMDA receptor and augment NMDA receptor-mediated excitatory postsynaptic potentials...Depending on the antibody concentrations, DNA–NMDA receptor antibodies can cause either neuronal dysfunction by transiently enhancing excitatory postsynaptic potentials or can result in neuronal cell death. This evidence could be of high relevance in terms of reversibility of symptoms...Furthermore, anti-brain antibodies were also shown to induce neuropsychiatric symptoms in patients with other autoimmune disorders such as celiac disease or inflammatory bowel diseases. Taken together, anti-brain antibodies were shown to cause neuropsychiatric pathology in different diseases presenting novel therapeutic options."
Inflammation disrupts neurogenesis
Both generation of new neurons and the support of synaptic health and plasticity are adversely affected by inflammation and this too is an avenue for treatment.
"Neurogenesis is a central mechanism required for neuronal maintenance and adaptive plasticity in the healthy and diseased brain. Inflammatory mediators have various effects on neurogenesis. Impairment of neurogenesis was shown in neurodegenerative diseases such as AD and neuropsychiatric disorders such as depression. Interestingly, approved AD drugs and chronic antidepressant treatment induce neurogenesis. Inflammation and microglial activation is detrimental for neurogenesis that can be restored by anti-inflammatory treatment. Moreover, microglia are not only involved in the maintenance of the neurogenic niche but also in synaptic maintenance. Of interest, systemic immune cells were shown to be involved in regulation of neurogenesis. CD4+ T-cells were shown to promote while CD8+ T-cells impair proliferation of neural progenitor cells...one may speculate that neuropsychiatric symptoms elicited by chronic inflammation may be driven by detrimental changes of neuronal homeostasis. Thus, specific immune modulatory treatment might be beneficial."
Inflammation is a core issue for brain health, cognition and mood
Case management of neuropsychiatric and neurodegenerative disorders requires discerning and treating the causes of chronic inflammation on an individual case basis. The authors conclude:
"Sustained systemic inflammation is a common feature of many autoimmune disorders, and is present in most sepsis survivors. Cognitive impairment is common in sepsis survivors, as well as patients suffering from chronic inflammatory conditions...Moreover, systemic inflammation occurring in a susceptible brain (e.g., patients with AD) may lead to even further disruption in quality of life and activities of daily living. Up to 95% of patients with SLE develop neuropsychiatric dysfunction...In patients with rheumatoid arthritis, the baseline vagal tone of is persistently low, suggesting a possible mechanism for persistent inflammation. Those examples indicate that the normal neuroimmune cross-talk in health can become deleterious during disease, particularly in a primed brain – one with preexistent damage. Recently, cellular, molecular, environmental, and genetic components have been linked to the persistent brain dysfunction of systemic inflammation. Here, we have discussed mechanistic evidence for the intricate interrelation between inflammation and neurodegeneration. Identification of druggable targets derived from these mechanisms holds the promise to prevent long-term disability and improve the quality of life in patients with chronic inflammatory conditions."
*Note: Transglutaminase-6 antibodies are included in the Wheat/Gluten Proteome Reactivity & Autoimmunity array from Cyrex Laboratories.